Thanks a lot for your prompt response!
Additionally, I’m assuming one doesn’t require do anything with free surfaces, right?
I have been able to run a 3D simulation and compare it against the same in 2D. Unfortunately, I haven’t been able to understand why the BC isn’t being applied. In my script I have
devsim.set_parameter(device="device", name=simple_physics.GetContactBiasName(new_name), value=bias)
simple_physics.CreateSiliconPotentialOnlyContact(device="device", region=zone, contact=new_name)
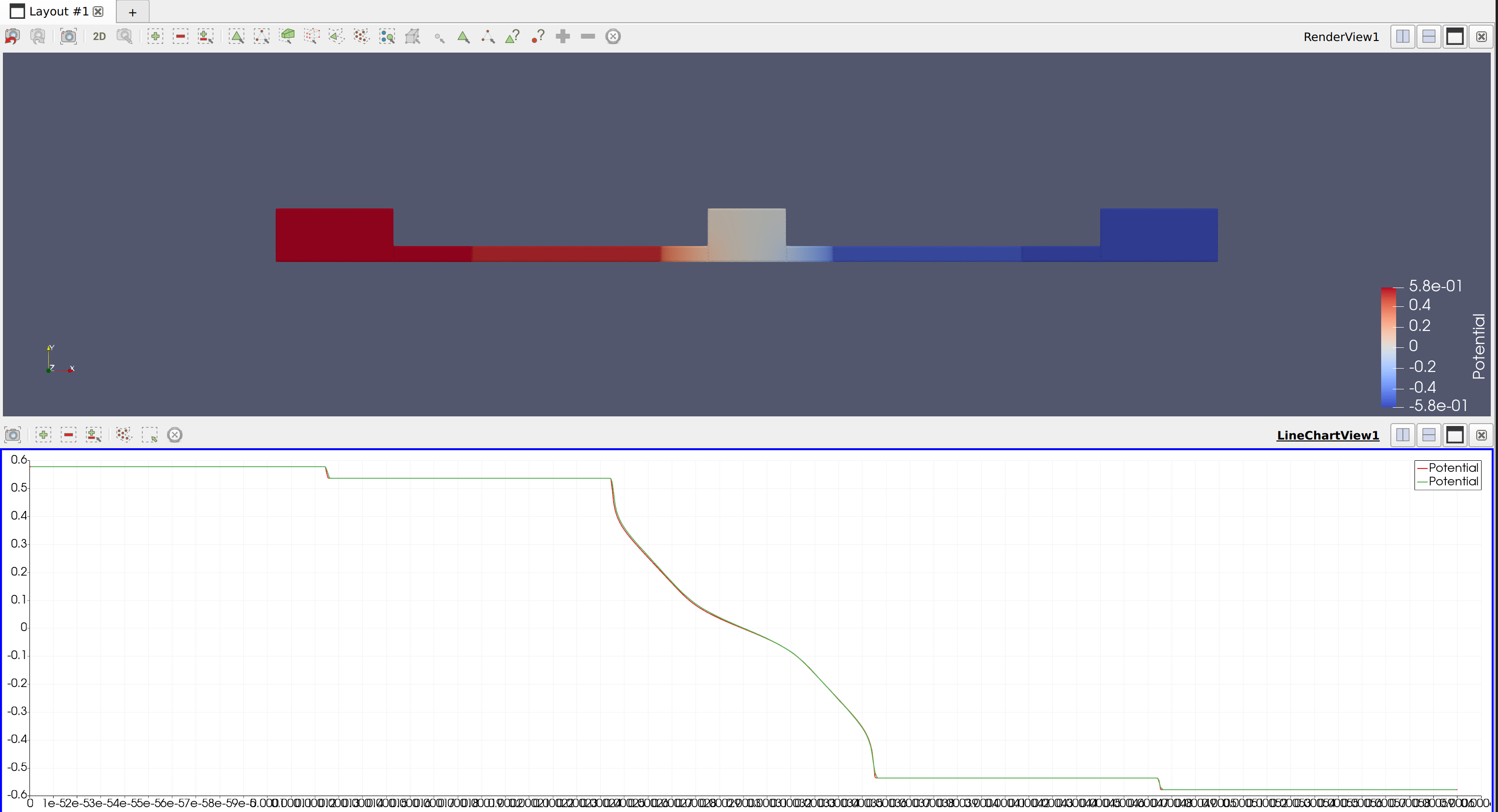
This is the result:
The lines in the figure are Potential for the 2D (brown) and 3D (green) cases. Both are very close to each other but they don’t comply with my bias, which I have set to 0.
I’m also including some output:
...
Region zone_8 has 1658 nodes.
Region zone_9 has 7699 nodes.
Contact zone_5_bc_5 in region zone_5 with 132 nodes
Contact zone_9_bc_9 in region zone_9 with 132 nodes
Adding interface zone_1_bc_0 with 33, 33 nodes
Adding interface zone_1_bc_1 with 33, 33 nodes
...
and later
Warning: Replacing equation with equation of the same name.
Region: zone_1, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_2, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_3, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_4, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_5, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_6, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_7, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_8, Equation: PotentialEquation, Variable: Potential
Warning: Replacing equation with equation of the same name.
Region: zone_9, Equation: PotentialEquation, Variable: Potential
Maybe the BC not being forced is the results of these warnings?